Introduction
SolexaQA calculates sequence quality statistics and creates visual representations of data quality for second-generation sequencing data. Originally developed for the Illumina system (historically known as “Solexa”), SolexaQA now also supports Ion Torrent and 454 data. Running directly on FASTQ files (now with support for compressed files too), the software has three component algorithms:
Analysis — the primary quality analysis and visualization tool. Designed to run on unmodified FASTQ files obtained directly from Illumina, Ion Torrent or 454 sequencers.
DynamicTrim — a read trimmer that individually crops each read to its longest contiguous segment for which quality scores are greater than a user-supplied quality cutoff (or alternately, the read segment returned by the BWA trimming algorithm).
LengthSort — a program to separate high quality reads from low quality reads. LengthSort assigns trimmed reads to paired-end, singleton and discard files based on a user-defined length cutoff.
DynamicTrim and LengthSort are typically used in combination to remove poor quality bases and/or reads from high throughput sequence data. Both programs should work on any FASTQ file (modified or unmodified, compressed or not).
The SolexaQA program automatically detects input FASTQ file formats (i.e., the Sanger, Illumina and Solexa formats described by Cock et al. 2010). It is designed to run on single-end or paired-end data, including reads from the latest versions of the HiSeq and MiSeq machines. High quality graphics are produced by interfacing automatically with R.
The SolexaQA software is freely released under the GNU General Public License version 3.
Citation
If you use this software, please cite:
Cox, M.P., D.A. Peterson, and P.J. Biggs. 2010. SolexaQA: At-a-glance quality assessment of Illumina second-generation sequencing data. BMC Bioinformatics 11:485. PDF, Web Link
Mailing List
We encourage users to join the (low traffic) Google Groups mailing list.
Members can discuss the SolexaQA program by sending emails to <solexaqa-users@googlegroups.com>. Help will be provided by the software developers, as well as by the user community.
Authors
SolexaQA was developed by Murray Cox, Patrick Biggs and Daniel Peterson. Release 2.0 was substantially reworked by Mauro Truglio. Release 3.0 was redeveloped in C++ by Mauro Truglio.
Please direct questions to Mauro Truglio <mauro.truglio@gmail.com>, Murray Cox <murray.p.cox@gmail.com> or Patrick Biggs <p.biggs@massey.ac.nz>.
Acknowledgments
SolexaQA++ uses Heng Li's kseq.h library to parse FASTQ files.
SolexaQA++ 
Release 3 is a complete redevelopment of the Perl-based SolexaQA 2 package in the programming language C++.
SolexaQA++ has undergone additional optimization, and the three separate components of the SolexaQA 2 suite (SolexaQA.pl, DynamicTrim.pl and LengthSort.pl) are now presented as options of a single, standalone program, which has been released for a variety of platforms. SolexaQA++ is no longer dependant on Perl, is cross-platform, and some functions are compatible with sequencing technologies other than Illumina, such as Ion Torrent and 454. All graphics are generated automatically using the award-winning graphical and statistical software R.
Changelog v.3.1.7.3:
Version 3.1.7.3: Minor update to improve presentation of results.
Changelog v.3.1.7.2:
Version 3.1.7.2: Minor bugfixes.
Changelog v.3.1.7.1:
Version 3.1.7.1: Bugfix for high-output NextSeq runs; minor bugfixes.
Changelog v.3.1.7:
Version 3.1.7: Added support for NextSeq reads.
Changelog v.3.1.6:
Version 3.1.6: Minor bugfixes.
Changelog v.3.1.5:
Version 3.1.5: Added subplot to histogram graph that shows the untrimmed length distribution; bugfixes.
Changelog v.3.1.4:
Bugfix in the analysis heatmap for variable length reads. In case a tile is entirely composed of reads shorter than all the other tiles, the squares for missing cycles are now painted gray.
Other minor bugfixes.
Changelog v.3.1.3:
Bugfix in the analysis histogram for variable length reads; fix for a malloc error on some systems when the -d option with a trailing forward slash is specified in dynamictrim; the summary graph for lengthsort shows the sample(s) name(s) as the graph title.
Changelog v.3.1.2:
Bugfix in the analysis matrix graph: it now contemplates ".fastq" files in which entire tiles are skipped, and names the tiles that are left correctly on the heat map.
Changelog v.3.1.1:
Bugfix in LengthSort (single-end mode)
Changelog v.3.1:
Corrected a file naming bug when operating on .gz files (Linux and Mac only); improved command line output;
Analysis: added warning message for SRA-generated fastq files not containing the tile number in the header;
DynamicTrim: added the [-a|--anchor] option, which allows trimming from the 3' end only;
LengthSort: New naming system for paired end reads:
- Read 1 are named "[read1 name].paired"
- Read 2 are named "[read2 name].paired"
- Single reads are named "[read1 name].single"
- Discarded reads are named "[read1 name].discard";
Changelog v.3.0:
Analysis (previously available as SolexaQA.pl): general performance improvements; the software can now process FASTQ files with variable read lengths, is compatible with Ion Torrent files (–t option), and .gz compressed files (Linux and OS X releases only). Minor cosmetic changes to graphs and command-line output have also been made.
DynamicTrim: general performance improvements (up to 100% faster); bug fix for truncated graphs on variable length reads; compatible with .gz compressed files (Linux and OS X releases only); new progress bar in command-line output.
LengthSort: paired-end mode includes a new option (–c) to remove non-matching reads from the two FASTQ files prior to processing; compatible with .gz compressed files (Linux and OS X releases only); new histogram output; new progress bar in command-line output.
Warnings:
-
On first use, Linux and OS X users may need to make the SolexaQA++ file executable ("Permission denied" error).
If so, use the command chmod +x SolexaQA++ - The use of compressed (.gz) FASTQ files slows down runtime, especially for large files.
- Due to library incompatibilities, the Windows version of SolexaQA++ does not support compressed files.
Requirements
SolexaQA++ works across a range of platforms, but is primarily designed for high performance UNIX machines. The program has been released as precompiled binaries for Linux, OS X and Windows, and can be compiled from the source code for other platforms.
SolexaQA++ requires a working (base) installation of R in your PATH in order to produce graphs. Most UNIX systems will already have this program installed. In Windows, R can be downloaded here and placed in the system's PATH by following this guide. No additional packages or modules are required.
Advanced users: to compile from source, SolexaQA++ requires the g++ compiler, together with the Boost and zlib libraries. Compilation instructions are included in the source code archive.
Usage Information
- Linux and OS X
-
Type the following commands in a console window:
./SolexaQA++ [command] [options] FASTQ_input_file(s)
- Windows
-
Open a console window (Start → type 'cmd' → Enter) and type:
SolexaQA++.exe [command] [options] FASTQ_input_file(s)
- Commands and options:
-
analysis SolexaQA++ analysis <FASTQ input file(s)> [-p|--probcutoff 0.05] [-h|--phredcutoff] [-v|--variance] [-m|--minmax] [-s|--sample 10000] [-b|--bwa] [-d|--directory path] [--sanger --illumina --solexa] [-t|--torrent]
OPTIONS:
-p or --probcutoff FLOAT probability value (between 0 and 1) at which base-calling error is considered too high (default: P = 0.05, or Q ≈ 13) or -h or --phredcutoff INT Phred quality score (between 0 and 41) at which base-calling error is considered too high -v or --variance calculate variance statistics (note: this approximately doubles the run time) -m or --minmax calculate minimum and maximum probabilities for each read position for each tile (note: this increases the run time by ~25%) -s or --sample INT integer number of sequences to be sampled per tile for statistics estimates (default: s = 10,000) -b or --bwa use BWA trimming algorithm -d or --directory STR path to directory where output files are saved (default: same as FASTQ files) --sanger Sanger FASTQ format (bypasses automatic format detection) --illumina Illumina FASTQ format (bypasses automatic format detection) --solexa Solexa FASTQ format (bypasses automatic format detection) -t or --torrent Ion Torrent or 454-generated FASTQ file
Quality cutoff values can be entered using either the -p or -h flags. If no quality cutoff value is given, SolexaQA defaults to P = 0.05. User-supplied Phred Q values are automatically converted to their corresponding probability value. All output is represented as probabilities of a base call being made in error. This is because interpretations of log probabilities can be problematic. For instance, consider difficulties interpreting the variance of log probabilities.
The user-supplied quality cutoff is used only to calculate the length of the best read segment; all other statistics are unaffected by this parameter.
Six output files are created per input FASTQ file. These have the name of the original FASTQ file with the following suffixes appended:
*.quality — a tab-delimited text file with mean probabilities of base call error for each read position (rows) for each tile (columns). The first column represents the global data (average base call error rate across all tiles). If variance, minimum, and maximum options are selected, these statistics are also displayed in the rightmost columns of this file.
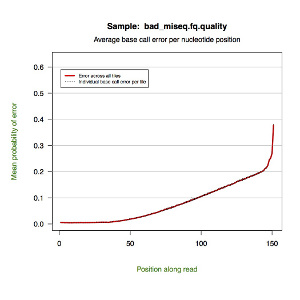
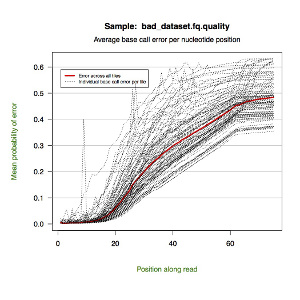
*.quality.pdf — a line graph showing the mean probability of error by nucleotide position along the read. Each tile is represented by a dotted line with a red line indicating the global mean.
*.matrix.pdf — a heat map depicting the mean probability of error for each nucleotide position and tile. Each row represents a different tile, and each column corresponds to a nucleotide position. The color scale progresses from white to black through yellow and orange, with pure white representing an error probability of 0 and pure black an error probability of 0.75 (i.e., any of the other three bases are equally likely to be correct).
*.matrix — a tab delimited text file showing mean probabilities of base calling error for each nucleotide position (columns) and each tile (rows). The arrangement of this matrix corresponds exactly to the layout of the heat map as shown in the *.pdf file.
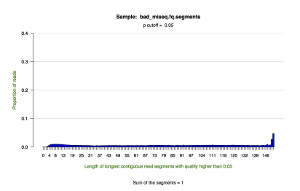
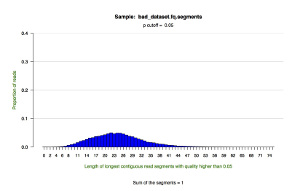
*.segments — contains metrics on the longest contiguous segment of each read in which the quality of each base is greater than the user-defined quality cutoff (defaults to P = 0.05). This tab delimited text file displays the proportion of all reads that contain a segment of a given length.
*.segments.hist.pdf — a histogram showing the distribution of the longest contiguous segment of each read in which the quality of each base is greater than the user-defined quality cutoff (defaults to P = 0.05). This figure draws upon the data in the *.segments file.
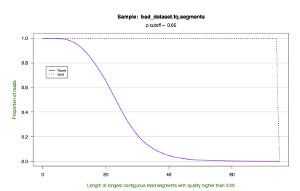
*.segments.cumulative.pdf — a line graph showing the cumulative frequency of trimmed read lengths. This figure also draws upon the data in the *.segments file.
dynamictrim SolexaQA++ dynamictrim <FASTQ input file(s)> [-p|--probcutoff 0.05] [-h|--phredcutoff] [-b|bwa] [-d|--directory path] [--sanger --illumina --solexa] [-t|--torrent]
OPTIONS:
-p or --probcutoff FLOAT probability value (between 0 and 1) at which base-calling error is considered too high (default: P = 0.05, or Q ≈ 13) or -h or --phredcutoff INT Phred quality score (between 0 and 41) at which base-calling error is considered too high -b or --bwa use BWA trimming algorithm -d or --directory STR path to directory where output files are saved (default: same as FASTQ files) --sanger Sanger FASTQ format (bypasses automatic format detection) --illumina Illumina FASTQ format (bypasses automatic format detection) --solexa Solexa FASTQ format (bypasses automatic format detection) -a or --anchor Reads will only be trimmed from the 3′ end -t or --torrent Ion Torrent or 454-generated FASTQ file
Quality cutoff values can be entered using either the -p or -h flags. If no quality cutoff value is given, DynamicTrim defaults to P = 0.05. User-supplied Phred Q values are automatically converted to their corresponding probability value.
Three output files are created per input FASTQ file. These have the name of the original FASTQ file with the following suffixes appended:
*.trimmed — a FASTQ file containing the trimmed reads.
*.segments — contains metrics on the longest contiguous segment of each read in which the quality of each base is greater than the user-defined quality cutoff (defaults to P = 0.05). This tab delimited text file displays the proportion of all reads that contain a segment of a given length.
*.segments.hist.pdf — a histogram showing the distribution of the longest contiguous segment of each read in which the quality of each base is greater than the user-defined quality cutoff (defaults to P = 0.05). This figure draws upon the data in the *.segments file.
lengthsort SolexaQA++ lengthsort <one single-end or two paired-end FASTQ files> [-l|--length 25] [-d|--directory path] [-c|--correct]
OPTIONS:
-l or --length INT length cutoff (default: 25 nucleotides) -d or --directory STR path to directory where output files are saved (default: same as FASTQ files) -c or --correct when running in paired-end mode, removes unpaired reads from the two fastq files, saves paired reads into two new *.fastq.clean files, and processes them as normal.
LengthSort assigns trimmed reads to files based on a user-defined length cutoff. Single-end data is sorted into reads that pass the length threshold (*.single) and reads smaller than the length threshold (*.discard). Paired-end data is sorted into two useable paired read files (*.paired1 and *.paired2), usable single reads (*.single) and non-usable reads that do not pass the length threshold (*.discard).
Either four or six output files are created per input FASTQ file. These have the name of the original FASTQ file with the following suffixes appended:
*.single — reads that are larger than or equal to the length cutoff. If paired-end data is being analyzed, these are reads that fulfill the length requirement, while their paired read does not.
*.discard — reads that are smaller than the length cutoff.
*.summary.txt — a tab delimited text file listing the number of reads assigned to the *.single and *.discard files (and if run on paired-end data, the *.paired1 and *.paired2 files as well).
*.summary.txt.pdf — a graphic representation of *.summary.txt using a barplot.
Paired-end data only:
[read1_name].paired — forward reads that are larger than or equal to the length cutoff. The reverse read pair also passes the length cutoff and is present in the [read2_name].paired file.
[read2_name].paired — reverse reads that are larger than or equal to the length cutoff. The forward read pair also passes the length cutoff and is present in the [read1_name].paired file.
SolexaQA 2 
Warning: SolexaQA 2 is legacy code as of August 2014. The software is still supported by the developers, but no further development is planned; all new development effort will be placed on SolexaQA++.
Release 2.0 is a substantial rewrite of the SolexaQA 1 package.
The software has been heavily optimized, and for most datasets, release 2.0 now runs 2-3 times faster than earlier versions. The graphical outputs have been completely reworked — they are now more informative than ever and more visually appealing. This version no longer requires a working installation of matrix2png. All graphics are generated automatically using the award winning graphical and statistical software R.
Release 2.1 is a minor code change. Heat maps are now returned as pdfs. This resolves issues writing png files using some versions of R under UNIX.
Release 2.2 is a minor code change. 'Empty' sequences can now be read by the BWA algorithm of DynamicTrim.
Release 2.3 handles a wider range of header lines, including the modified header lines used in the Sequence Read Archive (SRA).
Release 2.4 fixes a bug in the read sampling algorithm. The output is not changed, but this version now runs an order of magnitude faster on large files.
Release 2.5 fixes a bug that truncates results for HiSeq runs with 96 tiles.
Requirements
The SolexaQA package works across a range of platforms, but is primarily designed for high performance UNIX machines. The code works without modification on OS X.
SolexaQA needs working (base) installations of Perl and R. Most UNIX systems will already have these programs installed. No additional packages or modules are required.
Usage Information
- SolexaQA
-
perl SolexaQA.pl FASTQ_input_files [-p|probcutoff 0.05] [-h|phredcutoff] [-v|variance] [-m|minmax] [-s|sample 10000] [-b|bwa] [-d|directory path] [-sanger -illumina -solexa]
Command line arguments:
-p or -probcutoff # probability value (between 0 and 1) at which base-calling error is considered too high (default; P = 0.05, or Q ≈ 13) or -h or -phredcutoff # Phred quality score (between 0 and 41) at which base-calling error is considered too high -v or -variance calculate variance statistics (note: this approximately doubles the run time) -m or -minmax calculate minimum and maximum probabilities for each read position for each tile (note: the increases the run time by ~25%) -s or-sample # integer number of sequences to be sampled per tile for statistics estimates (default; s = 10,000) -b or -bwa use BWA trimming algorithm -d or -directory path to directory where output files are saved -sanger Sanger FASTQ format (bypasses automatic format detection) -illumina Illumina FASTQ format (bypasses automatic format detection) -solexa Solexa FASTQ format (bypasses automatic format detection)
Quality cutoff values can be entered using either the -p or -h flags. If no quality cutoff value is given, SolexaQA defaults to P = 0.05. User-supplied Phred Q values are automatically converted to their corresponding probability value.All output is represented as probabilities of base call error. This is because interpretations of log probabilities can be problematic. For instance, consider difficulties interpreting the variance of log probabilities.
The user-supplied quality cutoff is used only to calculate the length of the best read segment; all other statistics are unaffected by this parameter.
Six output files are created per input FASTQ file. These have the name of the original FASTQ file with the following suffixes appended:
*.quality — a tab-delimited text file with mean probabilities of base call error for each read position (rows) for each tile (columns). The first column represents the global data (average base call error rate across all tiles). If variance, minimum, and maximum options are selected, these statistics are also displayed in the rightmost columns of this file.
*.quality.pdf — a line graph showing the mean probability of error by nucleotide position along the read. Each tile is represented by a dotted line with a red line indicating the global mean.
*.matrix.pdf — a heat map depicting the mean probability of error for each nucleotide position and tile. Each row represents a different tile, and each column corresponds to a nucleotide position. The color scale progresses from white to black through yellow and orange, with pure white representing an error probability of 0 and pure black an error probability of 0.75 (i.e., any of the other three bases are equally likely to be correct).
*.matrix — a tab delimited text file showing mean probabilities of base calling error for each nucleotide position (columns) and each tile (rows). The arrangement of this matrix corresponds exactly to the layout of the heat map as shown in the *.pdf file.
*.segments — contains metrics on the longest contiguous segment of each read in which the quality of each base is greater than the user-defined quality cutoff (defaults to P = 0.05). This tab delimited text file displays the proportion of all reads that contain a segment of a given length.
*.segments.hist.pdf — a histogram showing the distribution of the longest contiguous segment of each read in which the quality of each base is greater than the user-defined quality cutoff (defaults to P = 0.05). This figure draws upon the data in the *.segments file.
*.segments.cumulative.pdf — a line graph showing the cumulative frequency of trimmed read lengths. This figure also draws upon the data in the *.segments file.
DynamicTrim-
perl DynamicTrim.pl FASTQ_input_files [-p|probcutoff 0.05] [-h|phredcutoff] [-b|bwa] [-d|directory path] [-sanger -illumina -solexa] [-454]
Command line arguments:
-p or-probcutoff # probability value (between 0 and 1) at which base-calling error is considered too high (default; P = 0.05, or Q ≈ 13) or -h or -phredcutoff # Phred quality score (between 0 and 41) at which base-calling error is considered too high -b or -bwa use BWA trimming algorithm -d or -directory path to directory where output files are saved -sanger Sanger FASTQ format (bypasses automatic format detection) -illumina Illumina FASTQ format (bypasses automatic format detection) -solexa Solexa FASTQ format (bypasses automatic format detection) -454 select this option if trimming Roche 454 or Ion Torrent data in FASTQ format (experimental feature) Quality cutoff values can be entered using either the -p or -h flags. If no quality cutoff value is given, DynamicTrim defaults to P = 0.05. User-supplied Phred Q values are automatically converted to their corresponding probability value.
Three output files are created per input FASTQ file. These have the name of the original FASTQ file with the following suffixes appended:
*.trimmed — a FASTQ file containing the trimmed reads.
*.segments — contains metrics on the longest contiguous segment of each read in which the quality of each base is greater than the user-defined quality cutoff (defaults to P = 0.05). This tab delimited text file displays the proportion of all reads that contain a segment of a given length.
*.segments.hist.pdf — a histogram showing the distribution of the longest contiguous segment of each read in which the quality of each base is greater than the user-defined quality cutoff (defaults to P = 0.05). This figure draws upon the data in the *.segments file.
Due to frequent requests, DynamicTrim is now able to trim 454 and Ion Torrent data if provided in FASTQ format. The trimming algorithm is functionally identical to that used for Sanger-style FASTQ data. Although 454 and Ion Torrent read trimming appears to work, we do not know how useful this feature will be. Trimming seems to occur most frequently at homopolymer repeats, which is where 454 and Ion Torrent data quality is typically most reduced. This function is currently provided as an experimental feature.
Note that SolexaQA, which works very differently to DynamicTrim, cannot accommodate 454 or Ion Torrent data. LengthSort can accommodate the output files produced by DynamicTrim on 454 or Ion Torrent data.
LengthSort-
perl LengthSort.pl one single-end or two paired-end FASTQ files [-l|length 25] [-d|directory path]
Command line arguments:
-l or -length length cutoff [defaults to 25 nucleotides] -d or -directory path to directory where output files are saved LengthSort assigns trimmed reads to files based on a user-defined length cutoff. Single-end data is sorted into reads that pass the length threshold (*.single) and reads smaller than the length threshold (*.discard). Paired-end data is sorted into two useable paired read files (*.paired1 and *.paired2), usable single reads (*.single) and non-usable reads that do not pass the length threshold (*.discard).
Either three or five output files are created per input FASTQ file. These have the name of the original FASTQ file with the following suffixes appended:
*.single — reads that are larger than or equal to the length cutoff. If paired-end data is being analyzed, these are reads that fulfill the length requirement, while their paired read does not.
*.discard — reads that are smaller than the length cutoff.
*.summary.txt — a tab delimited text file listing the number of reads assigned to the *.single and *.discard files (and if run on paired-end data, the *.paired1 and *.paired2 files as well).
Paired-end data only:
*.paired1 — forward reads that are larger than or equal to the length cutoff. The reverse read pair also passes the length cutoff and is present in the *.paired2 file.
*.paired2 — reverse reads that are larger than or equal to the length cutoff. The forward read pair also passes the length cutoff and is present in the *.paired1 file.
Example MiSeq Figures
| Heat Map |  |
 |
| Read Quality | 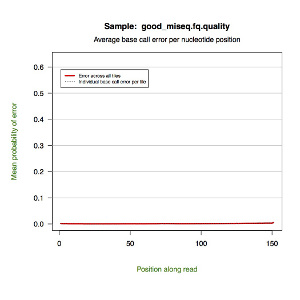 |
 |
| Length Histogram | 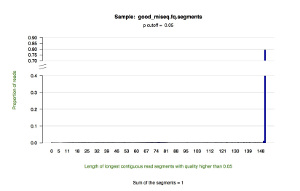 |
 |
| Cumulative Length | 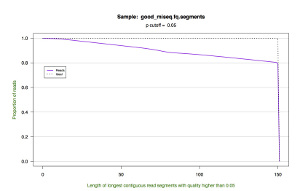 |
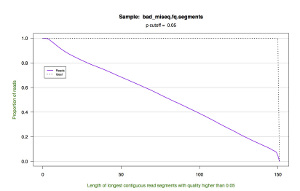 |
Example GAIIx Figures
| Heat Map | 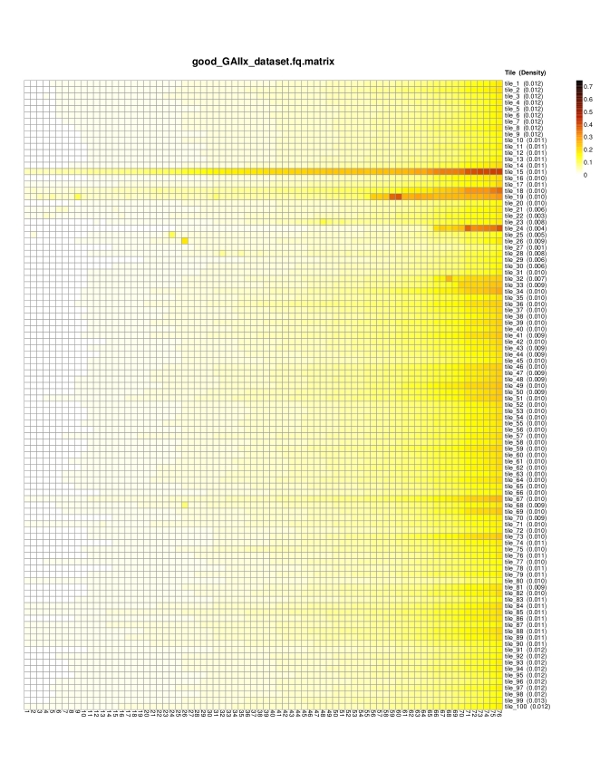 |
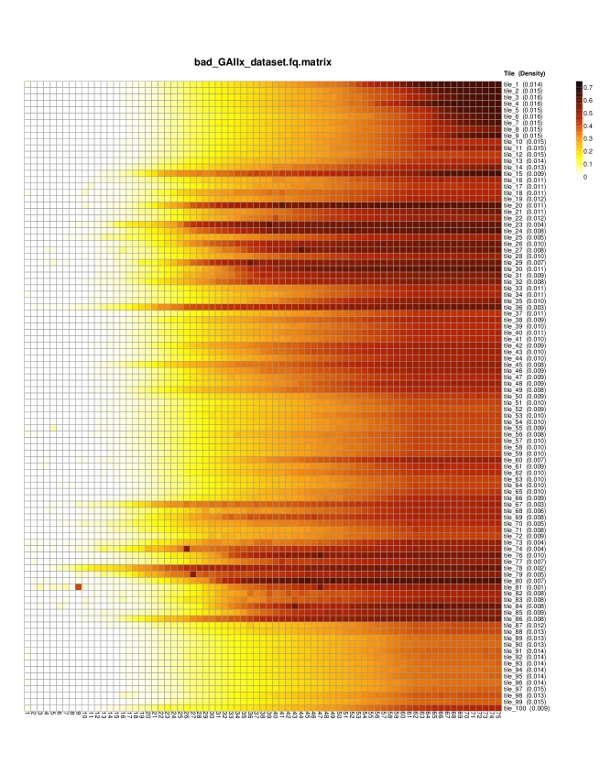 |
| Read Quality | 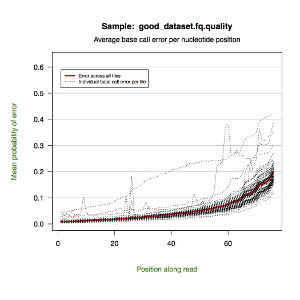 |
 |
| Length Histogram | 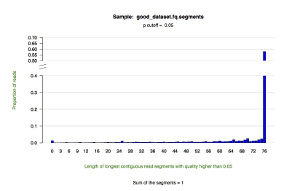 |
 |
| Cumulative Length | 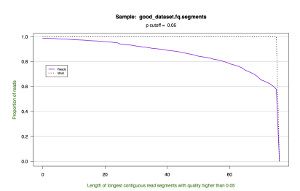 |
 |
Frequently Asked Questions
- How does the SolexaQA software trim reads?
- How does the SolexaQA software compare to other trimming methods?
- Can the SolexaQA software handle paired-end data?
- How does DynamicTrim deal with paired-end data?
- Can the SolexaQA software use Illumina 1.5+ and 1.8+ pipeline data?
- Can the SolexaQA software be used on 454 and Ion Torrent data?
- Can the SolexaQA software handle different header line formats?
- Is there a public repository (e.g. Git) for SolexaQA?
- How does SolexaQA trim reads?
-
By default, SolexaQA extracts the longest contiguous sequence where all bases have a quality score higher than the user-defined threshold (defaults to P = 0.05, or equivalently, Q = 13).
Consider the following read with a poorly called 3′ base:
Base A C A C C T G C C G Quality score 30 30 30 30 30 30 30 30 30 10
Using its default threshold, the SolexaQA software would trim this read to:
Base A C A C C T G C C Quality score 30 30 30 30 30 30 30 30 30
Now consider a read also containing a poorly called internal base:
Base A C A C C T G C C G Quality score 30 10 30 30 30 30 30 30 30 10
Using its default threshold, the SolexaQA software would trim this read to:
Base A C C T G C C Quality score 30 30 30 30 30 30 30
How does the SolexaQA software compare to other trimming methods?-
Most trimming algorithms are poorly documented. However, one alternative approach is implemented in the read mapper BWA.
For any given read, BWA maximizes the following equation:
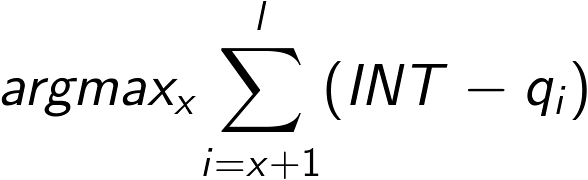
if ql < INT where l is the original read length and INT is a user-defined quality threshold (Q score).
So how does this differ from the SolexaQA software? As far as we can tell, BWA and the SolexaQA software trim approximately the same way for reads with poorly called 3′ bases. Consider the read used as an example above:
Base A C A C C T G C C G Quality score 30 30 30 30 30 30 30 30 30 10 For this read, the BWA equation is maximized at x = 10. BWA will therefore remove the final base, exactly the same as the SolexaQA software:
Base A C A C C T G C C Quality score 30 30 30 30 30 30 30 30 30 However, now consider the read containing an internal error:
Base A C A C C T G C C G Quality score 30 10 30 30 30 30 30 30 30 10
The BWA equation is still maximized at x = 10. BWA will therefore trim off only the final base, leaving the poor quality base at position 2 intact:
Base A C A C C T G C C Quality score 30 10 30 30 30 30 30 30 30 The BWA algorithm has a number of distinguishing characteristics:
- Reads can only be trimmed from the 3′ end.
- Error prone base calls may be retained.
- Calculations are performed on quality scores (i.e., log probabilities of error). Summing these log error values is the equivalent of multiplying the actual probabilities. These two measurements are not linearly related, and summing the log values therefore places increasingly less weight on poorly called bases.
In comparison, the SolexaQA software:
- Permits trimming from either end of the read.
- Removes all bases with qualities worse than the user-defined threshold.
- Trims on actual probabilities of error (not their log values).
The trimming approach implemented in the SolexaQA software appears more conservative than that of BWA (e.g., it removes all poor quality bases). Whether this suits your needs will largely depend on exactly what analyses you want to do with your trimmed data.
Dynamic trimming appears to improve read mapping rates, as well as improving validation rates of SNP calls. Because de novo assemblers have their own error correction mechanisms, the effect of read trimming on assembly quality appears to be strongly dataset dependent. We encourage robust testing of all methods on both trimmed and untrimmed data.
Can the SolexaQA software handle paired-end data?-
Yes, the SolexaQA software was specifically designed for paired end data. Just supply multiple FASTQ files on the command line.
How does DynamicTrim deal with paired-end data?-
This question only arises when one of the reads is so bad that it has to be removed. We opt to use dummy reads, as described in the following example:
Before trimming:
Forward read file:
@GATC-23_0002:1:1:1142:10040#0/1
GGCTGTTATATATTAGTGATATTTTGGCA
+GATC-23_0002:1:1:1142:10040#0/1
^aaa^a`ab`fffffffddddddddabBB
Reverse read file:
@GATC-23_0002:1:1:1142:10040#0/2
TTGTGTGGTCTTTCAGAAGCTGGGAAACC
+GATC-23_0002:1:1:1142:10040#0/2
BBBBBBBBBBBBBBBBBBBBBBBBBBBBB
After trimming:
Forward read file:
@GATC-23_0002:1:1:1142:10040#0/1
GGCTGTTATATATTAGTGATATTTTGG
+GATC-23_0002:1:1:1142:10040#0/1
^aaa^a`ab`fffffffddddddddab
Reverse read file:
@GATC-23_0002:1:1:1142:10040#0/2
N
+GATC-23_0002:1:1:1142:10040#0/2
B
The dummy read in the reverse read file maintains read order between the forward and reverse read files. This seems preferable to i) having different numbers of unpaired reads in the forward and reverse files, or ii) leaving empty lines in the output file, which many downstream applications are not able to handle.
In terms of downstream applications, de novo assembly software like ABySS or Velvet will simply exclude any reads smaller than the k-mer, so these output files can be used as they are. Alternately, LengthTrim will sort reads into files based on a user-defined cutoff value (say, reads no smaller than 25 nucleotides). Which of these options you choose to employ will depend on the exact nature of your study.
Can the SolexaQA software use Illumina 1.5+ and 1.8+ pipeline data?-
Yes. Some sources suggest that the Illumina 1.5+ pipelines have redefined the use of quality scores 0 to 2. Values 0 and 1 are no longer used. Value 2, encoded by ASCII value 66 (i.e., character “B”), is used only at the end of reads as a Read Segment Quality Control Indicator. Consequently, it is suggested that the “B” character should not be treated as a quality score.
However, we believe that there is an alternative interpretation of these low quality scores that works equally well.
Imagine that we have no information about a nucleotide position, and thus, we assign it a base randomly (say, G). The probability that this base really is a G is 0.25. The probability that it is not a G is therefore 0.75. It is these probabilities of error that are recorded in FASTQ files as ASCII characters.
However, at these high rates of error, ASCII characters encode error probabilities only very sparsely:
ASCII character Quality score Error probability @ 0 1.000 A 1 0.794 B 2 0.631 C 3 0.501 ⋮ ⋮ ⋮ A randomly assigned base should presumably be assigned the ASCII character “B” because probabilities of error greater than 0.75 make little sense and we would not expect to observe them. Indeed, given that the “@” character is used in FASTQ header lines, Illumina has a very good reason to exclude them.
In short, regardless of the details of the 1.5+ variant of Illumina quality scores, the ASCII characters (and associated quality scores) still seem to retain their natural interpretation — as actual probabilities of error. Until this situation is clarified, we have decided not to make any alterations to the SolexaQA code.
In the 1.8+ pipeline, Illumina have extended their quality scores from 40 to 41. The Phred scores retain their natural interpretation, and the SolexaQA software can accommodate these larger Q values.
Can the SolexaQA software be used on 454 and Ion Torrent data?-
Yes, SolexaQA++ is fully compatible with 454 and Ion Torrent using the -t option.
As of June 2011, both 454 and Ion Torrent data have been made available in Sanger FASTQ format, and can therefore be processed.
BGI has released example Ion Torrent data for Escherichia coli isolate TY-2482. Here are untrimmed and trimmed datasets for comparison.
The following graph shows the distribution of read lengths before (black) and after (red) dynamic read trimming (P = 0.05). Note that early release Ion Torrent data is still subject to relatively high error rates, particularly around homopolymer repeats.
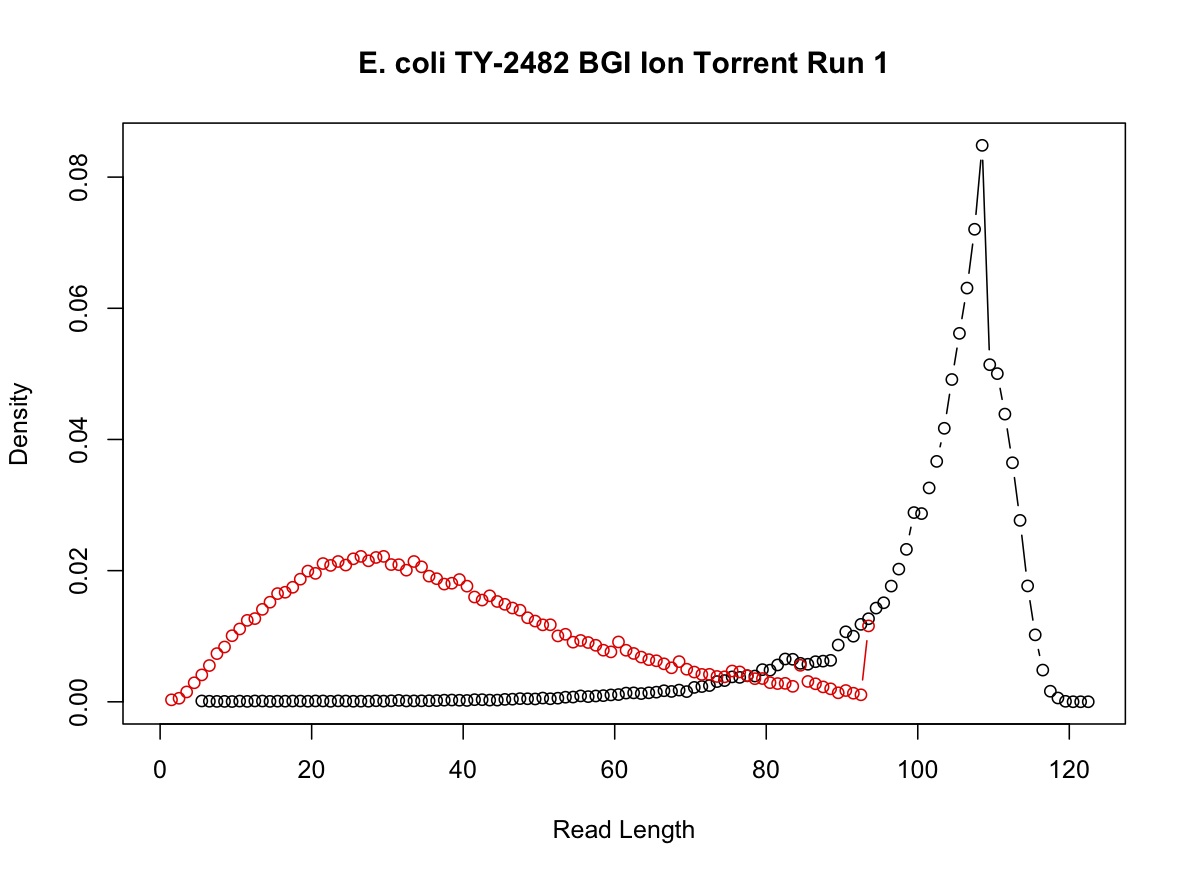
- Can the SolexaQA software handle different header line formats?
-
Yes, SolexaQA is being continually updated to handle different file formats released by Illumina, including changes in the header lines.
A major change occurred with the release of Illumina’s CASAVA 1.8. Header lines that formerly looked similar to this:
@EAS139-23_0002:1:100:1142:10040#ATCACG/1
changed to something like this:
@EAS139:136:FC706VJ:2:2104:15343:197393 1:Y:18:ATCACG
As of CASAVA 1.8, Illumina has also released data solely in Sanger FASTQ format.
Note that Illumina is perversely fond of changing its read header lines (and number of tiles), often for no obviously good reason. Other groups, such as the Sequence Read Archive (SRA), have also modified the header lines. To date, we have encountered — and changed the SolexaQA code to accommodate — the following header line variants:
@AGRF-23_0002:1:100:1142:10040#0/1
@114222772_110210mm_lane7.txt_SN603_WA003:7:1:2036.50:19999.80#0/1
@114222772_110210mm_lane7:7:1:2036.50:19999.80#0/1
@txt_SN603_WA003:7:1:2036.50:19999.80#0/1
@7.t:7:1:2036.50:19999.80#0/1
@HWUSI-EAS1758R:18:62RU0AAXX:5:1:1030:952 1:N:0:
@EAS139:136:FC706VJ:2:1101:15343:197393 1:Y:18:ATCACG
@SRR001666.1 071112_SLXA-EAS1_s_7:5:1:817:345 length=36
@HWI-ST938:49:B01D4ACXX:5:1101:2169:194234
@HWI-EAS209_0025_FC427:6:1:1041:14884#ACAGTG/2
@M00143:1:000000000-A0AH7:1:1:16598:1320 1:N:0:1
@8:1:670:774
@USI-EAS45-42B86AAXX:7:1:1793:1105:0:1:0
@SRR797058.1 HWI-ST600:227:C0WR4ACXX:7:1101:16297:2000/1We do not guarantee that SolexaQA can read all header lines out there, but it can handle the variants that we are aware of. If you come across another header format, please contact the developers so that it can be incorporated into future releases.
Is there a public repository (e.g. Git) for SolexaQA?-
Yes, SolexaQA's SourceForge page contains a "code" section (link) with the latest source code available.